Prokaryotes employ the abortive infection (Abi) strategy to curb phage propagation at the cost of individual cell death, with type II toxin–antitoxin (TA) systems serving as one of the primary weapons that execute this strategy. In parallel, the type VI CRISPR–Cas13 system also triggers abortive infection through RNA targeting and subsequent nonspecific cleavage. In recent years, genomic evidence has indicated that type II TA systems and CRISPR–Cas systems are frequently adjacent on the genome, suggesting a potential functional association between them. However, whether this physical co‑localization reflects a direct functional coupling has not been experimentally demonstrated until now.

Systematic genomic analysis of bacteria containing CRISPR–Cas13a revealed that, in some strains, a CRISPR array is interposed between hicA and hicB, giving rise to a distinctive hicA–CRISPR–hicB–cas13a–CRISPR architecture, implying a potential synergistic interplay between the two systems.
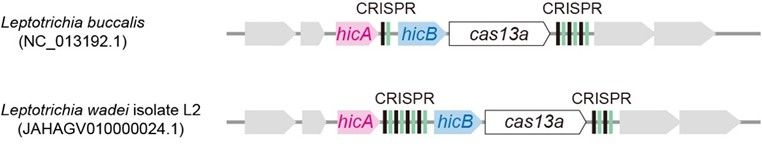
Gene arrangement features of the CCTA system
Further investigation revealed that HicB and Cas13a can form a complex both in vitro and in vivo. When expressed individually, neither protein inhibited E. coli growth; however, co‑expression significantly suppressed bacterial growth and conferred broad‑spectrum anti‑phage activity, accompanied by massive degradation of rRNA, tRNA, and mRNA. Mutating the catalytic site of Cas13a did not abolish RNA degradation or phage resistance, whereas simply increasing the expression level of HicB alone produced similar effects, indicating that HicB is the direct executor of RNA degradation.
Crystal structures showed that HicB exists as a tetramer, with lysine residues at positions 102, 103, and 104 of its C‑terminus serving as critical active sites. The structure of the Cas13a–HicB complex further revealed that two Cas13a molecules arrange the HicB tetramer in a tight sandwich‑like manner, bringing the key lysine residues into close spatial proximity, thereby activating the RNA‑degrading activity of HicB.
In the classical HicAB system, HicB inhibits the toxicity of HicA by binding to its active centre. The present study confirmed that HicA has a higher affinity for HicB than does Cas13a, and can competitively inhibit the Cas13a‑mediated activation of HicB, ensuring that the system is not aberrantly triggered under resting conditions.
Electrophoretic mobility shift assays (EMSA) and microscale thermophoresis (MST) experiments demonstrated that HicA, HicB, and the HicAB complex all bind to crRNA, with HicAB exhibiting the highest affinity. Cryo‑electron microscopy structures revealed that two crRNA molecules assemble with two HicAB heterohexamers into a hetero‑tetradecamer, in which the stem‑loop structure of crRNA is recognized and bound by HicA, neutralizing its catalytic activity. This discovery highlights that the system possesses hybrid features of both type II (proteinaceous antitoxin) and type III (RNA‑based antitoxin) TA systems.
The research team named this integrated system the CCTA system (CRISPR‑Cas–toxin‑antitoxin systems). This study breaks through the traditional view that antitoxins are limited to “inhibitory” functions, and reveals that Cas13a can directly bind to and reprogram the antitoxin HicB, conferring upon it RNA‑degrading activity and synergistically triggering abortive infection to block phage propagation. Meanwhile, the toxin HicA acts as a competitive inhibitor to prevent aberrant activation of HicB, and crRNA also serves an antitoxin function. Together, these findings provide the first description of a multi‑layered immune regulatory network coordinated by Cas13a, HicB, HicA, and crRNA. Furthermore, the binding mode between HicAB and crRNA offers a structural basis and new design principles for the development of RNA‑guided nucleic acid‑targeting tools.
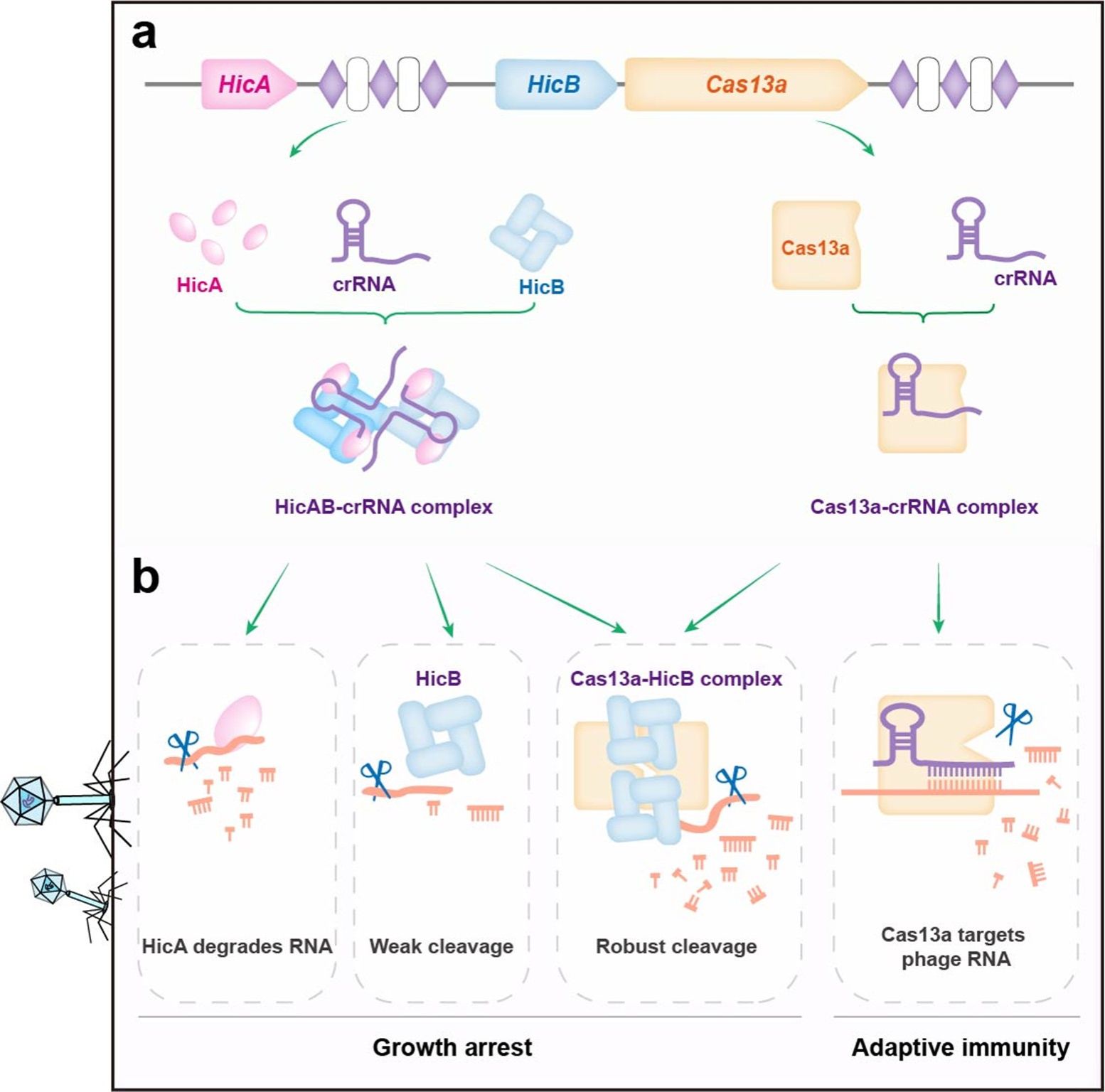
The multi‑strategy defense model of the CCTA system
The co‑corresponding authors of this paper are Professor Liu Liang and Associate Professor Chen Jiyun from the School of Life Sciences, Xiamen University, and Dr. Li Xueyan from the MRC Laboratory of Molecular Biology; the co‑first authors are Associate Professor Chen Jiyun, doctoral students Huang Linglong (2021 cohort) and Chen Hong (2023 cohort) from the School of Life Sciences, Xiamen University, and Dr. Li Xueyan from the MRC Laboratory of Molecular Biology, while postdoctoral fellows Lin Xiaofeng and Chen Ying, doctoral students Guo Chenmin and Fu Guowei, and Master's student Liu Xi, all from the School of Life Sciences, participated in the research. This work was supported by the National Natural Science Foundation of China, the Fujian Provincial Natural Science Foundation of China, the Fundamental Research Funds for the Central Universities, and the Scientific Research Foundation of State Key Laboratory of Vaccines for Infectious Diseases, Xiang An Biomedicine Laboratory.
Original article link: https://www.nature.com/articles/s41467-026-74930-z