吴乔课题组发文阐明扰动血红素代谢稳态诱导细胞焦亡的新机制
日期: 2025-12-24 访问数:细胞焦亡是由gasdermin家族蛋白介导的程序性死亡。在凋亡耐受的黑色素瘤细胞中高表达GSDMC,而在正常组织中低表达GSDMC,提示激活GSDMC诱导焦亡可能是治疗黑色素瘤的潜在方向。血红素是一种含铁的环状四吡咯化合物,在生命各种重要过程中发挥核心作用,包括电子传递、昼夜节律、氧化还原反应和基因表达等。血红素合成通常被认为是肿瘤存活的先决条件,许多类型的肿瘤在发生发展过程中伴随着血红素合成通路的上调。细胞命运的决定与自身代谢稳态密切相关,然而细胞内的代谢失衡信号如何转化为焦亡信号目前还不清楚。

近日,吴乔教授课题组在Signal Transduction and Targeted Therapy杂志发表题为《Disruption of heme homeostasis by nuclear receptor Nur77 induces pyroptosis through granzyme B-dependent GSDMC cleavage》的研究论文。该研究从机制上阐述了血红素代谢稳态失衡通过OPA1-ISR-granzyme B-GSDMC信号轴介导的细胞焦亡新通路。
前期,吴乔教授课题组揭示了代谢产物α-KG激活caspase-8剪切GSDMC诱导细胞焦亡的分子机制(Cell Research,2021)。为了进一步解析代谢应激激活GSDMC的分子机制,课题组在自己构建的化合物库中对2371个化合物进行筛选,最终发现小分子化合物DdBIC可以有效诱导多种肿瘤细胞发生GSDMC依赖的焦亡。机制研究表明线粒体电子传递链的SDH复合物中血红素匮乏是DdBIC调控GSDMC的上游起始信号。DdBIC靶向核受体Nur77后诱导其转运到线粒体,通过与SDHA互作提高SDH复合物活性,导致上游代谢物琥珀酰辅酶A的匮乏,损害血红素的从头合成途径。SDH复合物中的血红素具有防止电子泄露的关键功能。DdBIC处理导致SDH复合物中血红素的匮乏,由此引起电子泄露和mito-ROS提高。进而线粒体蛋白酶OMA1通过氧化修饰感知mito-ROS信号后,促使下游OPA1被切割成S-OPA1,并随着mPTP开放和线粒体外膜破裂释放到胞浆。S-OPA1与PERK互作进一步激活ISR(integrated stress response),后者再激活胞浆中的granzyme B活性,最终剪切GSDMC诱导黑色素瘤细胞发生焦亡性死亡。小鼠模型中DdBIC显著下调肿瘤内的血红素水平,通过Nur77和GSDMC介导的细胞焦亡有效地抑制肿瘤生长,无明显的毒副作用。DdBIC还能够有效激活抗肿瘤免疫,联用anti-PD-1抗体显著增强抗肿瘤的治疗效果。
综上,该研究不仅鉴定出全新的GSDMC依赖的焦亡诱导剂,发现肿瘤细胞来源的granzyme B的新功能,还阐明血红素代谢稳态失衡导致细胞焦亡的新通路,提示通过调节血红素水平可以拓宽治疗窗口,开辟治疗新策略。
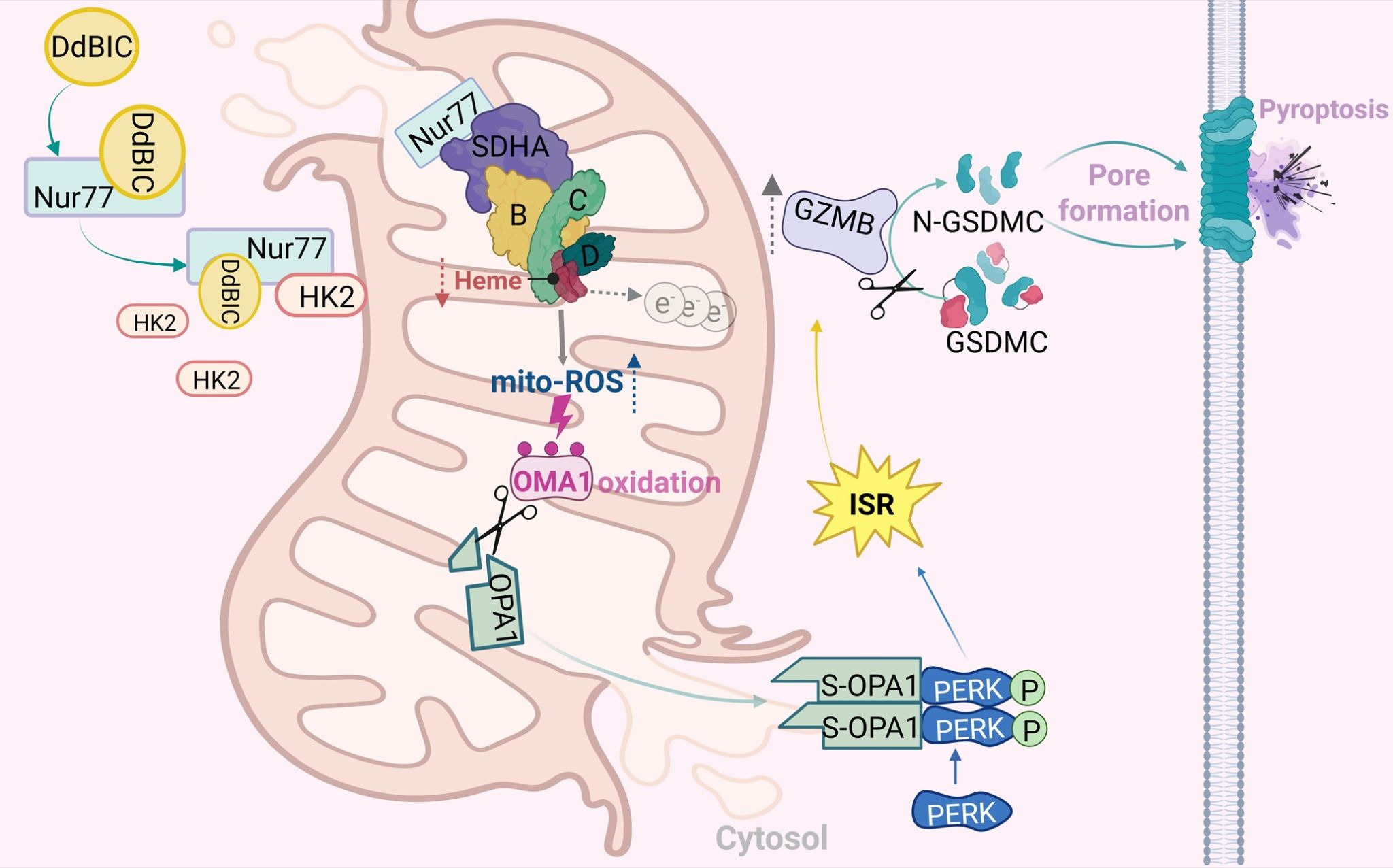
Nur77通过扰动血红素稳态诱导granzyme B-GSDMC介导的细胞焦亡模式图
吴乔教授、陈航姿教授、厦门大学附属第一医院陈学勤副教授和厦门大学药学院李福男副教授为本文的共同通讯作者。博士生吴流政、黄雅莹,硕士生胡海婧,厦门大学附属第一医院洪文斌博士后和厦门大学药学院博士生颜晗为本文的共同第一作者。该研究得到国家自然科学基金区域联合重点项目和重大研究计划重点支持、科技部重点研发计划,福建省自然科学基金和厦门大学汪德耀优秀研究生奖学金等项目的资助。
论文链接:https://www.nature.com/articles/s41392-025-02528-w
(图/文 吴乔课题组)
